Cancer de l’ovaire
Cancer testiculaire/para-testiculaire
Cancer vagin/utérus
Cancer vessie/prostate
CORTICOSURRENALOME
Cancer du rein
- Background
- Définition
Les cancers du rein regroupent un ensemble de tumeurs malignes développées à partir du parenchyme rénal. Bien que les types histologiques varient selon l’âge, 80% des cas sont des néphroblastomes (tumeurs de Wilms).
- Épidémiologie
-Rare : 7-8% des cancers de l’enfant
-Différents type histologique
Néphroblastome (TW): Le plus fréquent, 85%
Carcinome rénale (CR): en deuxième position, 5%
Sarcome à cellule clair (SCC) : en troisième position, 3%
Néphrome mésoblastique (NM): 3%
Tumeur rhabdoïde (TR): 2%
Autres : 2%
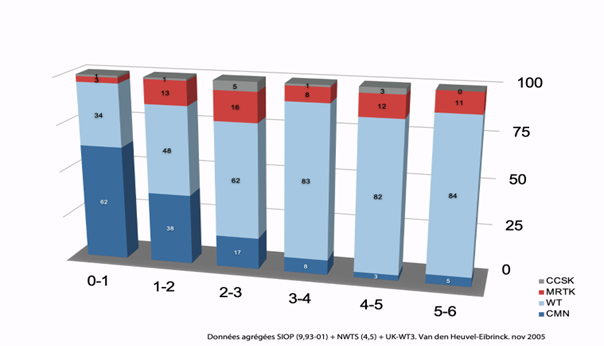
Type histologique varie selon l’âge
Age | Fréquent | Possible | Rare |
Naissance | NM | TW | TR |
< 1 an | TW, NM | TR,SCC | |
1 – 5 ans | TW | SCC | NM (<3 ans), TR |
5 – 10 ans | TW | SCC, CR | |
11-15 ans | TW, CR |
- Particularités
-Contexte génétique dans 10% des cas
Syndrome de WAGR (tumeur de Wilms, Aniridie, anomalies Génito-urinaire, Retard mental)
Syndrome de Denys-Drash (sclérose mésangiale diffuse, anomalies du développement sexuel)
Syndrome de Wiedmann Beckwith (hémihypertrophie, macroglossie, omphalocèle, viscéromégalie)
Syndrome de Simpson-Golabi-Behmel (macrosomie, dysmorphie faciale, malformations vertébrales, anoamlie membres, viscéromégalie, atteinte génitale, déficience intellectuelle)
Syndrome de Perlman
Syndrome de Sotos
- Atteinte rénale propre
- Risque de tumeur controlatérale :Surveillance tous les 3-4 mois pendant 7 ans
- Rappels histologiques
2.1) Néphroblastome
Tumeur associant dans des proportions variables d’une composante épithéliale, d’une composante mésenchymateuse ou stromale et d’une composante blastémateuse +/- Anaplasie (focale, diffuse).
Classification de l’International Society of Pediatric Oncology (SIOP)
- Histologie favorable :
- Tumeur stromale : Prédominance de tissu conjonctif.
- Tumeur épithéliale : Prédominance de structures tubulaires et glomérulaires.
- Tumeur mixte : Composée de tissus stromal et épithélial en proportions variables.
- Histologie défavorable :
- Blastème prédominant : Présence majeure de cellules blastemales, résistant à la chimiothérapie.
- Anaplasie : Caractérisée par une augmentation de la taille nucléaire, un polymorphisme nucléaire, et une hyperchromasie. L’anaplasie peut être focale ou diffuse.
- Anaplasie focale : Zones d’anaplasie limitées et localisées.
- Anaplasie diffuse : Zones d’anaplasie étendues et généralisées.
Classification de l’International Society of Pediatric Oncology (SIOP) et de la Children’s Oncology Group
STADE | COG | SIOP |
I | Tumeur limitée au rein, complètement réséquée Capsule rénale intacte Pas de rupture ni de biopsie Pas de métastases nodales ou hématogènes. | Tumeur limitée au rein, complètement réséquée. Tumeur présente dans la graisse périrénale mais entourée par une (pseudo)capsule fibreuse Tumeur peut montrer une croissance protrusive dans le bassinet ou l’uretère sans infiltrer leurs parois. Pas d’implication des vaisseaux ou des tissus mous du sinus rénal Pas de rupture ou de biopsie avant l’ablation. |
II | Tumeur dans la graisse périrénale mais complètement réséquée. Tumeur infiltre le sinus rénal ou les vaisseaux sanguins et lymphatiques en dehors du parenchyme rénal mais est complètement réséquée. Tumeur infiltre les organes adjacents ou la veine cave mais est complètement réséquée. | Tumeur viable est présente dans la graisse périrénale sans être recouverte par une (pseudo)capsule mais est complètement réséquée (les marges de résection sont saines). Tumeur viable infiltre les tissus mous du sinus rénal. Tumeur viable infiltre les vaisseaux sanguins ou lymphatiques du sinus rénal ou du tissu périrénal. Tumeur viable infiltre la paroi du bassinet ou de l’uretère. Tumeur viable infiltre la veine cave ou les organes adjacents (sauf la glande surrénale) mais est complètement réséquée. |
III | Tumeur résiduelle ou métastases non hématogènes confinées à l’abdomen. Ganglions lymphatiques abdominaux atteints. Implants péritonéaux tumoraux. Rupture de la tumeur avant ou pendant la chirurgie. Résidu tumoral macroscopique dans l’abdomen. Biopsie de la tumeur (y compris la biopsie à l’aiguille fine) avant l’ablation du rein. Marges de résection envahies par la tumeur ou la tumeur est sectionnée pendant la résection (excision en morceaux). | Tumeur viable est présente aux marges de résection. Ganglions lymphatiques abdominaux contiennent une tumeur viable ou non viable. Thrombus viable ou non viable présent aux marges de résection de l’uretère, de la veine rénale ou de la veine cave inférieure. Thrombus tumoral viable ou non viable dans la veine cave inférieure retiré en morceaux par un chirurgien. Rupture tumorale préopératoire ou peropératoire, confirmée par examen microscopique (tumeur viable à la surface du spécimen au niveau de la rupture). Biopsie en coin ou ouverte avant chimiothérapie ou chirurgie préopératoire. Implants tumoraux (viables ou non viables) dans l’abdomen. La tumeur (viable ou non viable) a pénétré la surface péritonéale. |
IV | Métastases hématogènes ou dissémination au-delà de l’abdomen. | Métastases hématogènes (poumon, foie, os, cerveau) ou métastases ganglionnaires en dehors de la région abdomino-pelvienne. |
V | Tumeurs bilatérales au diagnostic ; chaque côté doit être sous-classé selon les critères ci-dessus. | Tumeurs bilatérales au diagnostic ; chaque côté doit être sous-classé selon les critères ci-dessus. |
- Néphrome mésoblastique
Tumeur conjonctive de bas grade avec 3 types histologiques
-Classique (leiomyome-like) = fibromatose infantile rénale
-Cellulaire = fibrosarcome infantiler rénal (Transcrit de fusion ETV6-NTRK3 / EML4–NTRK3)
-Mixte
- Tumeur rhabdoïde
-Tumeur présentant des cellules rhabdoïdes (inclusions éisonophiles intra-cytoplasmiques, noyau volumineux excentré, nucléole proéminent) avec une densité cellulaire forte et des nappes de cellules non cohésives.
-Immunohistochimie : expression de vimentine, cytokératines, EMA, S-100, Desmine, Myoglobine
-Biologie moléculaire : Perte d’expression d’INI1/SMARCB1
- Carcinome rénal
Tumeur d’architecture papillaire avec des microcalcifications
Biologie moléculaire : CK +/- , CD10++, PAX2++, PAX8++, TFE3 ou TFEB : – TFE3 : Melan A et HMB45 +/-, TFE3 +++
- Sarcome à cellule claire
Nids/cordons de cellules tumorales séparés par des septa fibrovasculaires branchés
Immunohistochimie : vimentine, Bcl2, Cycline D1, BCOR
Biologie moléculaire : Transcrit de fusionYWHAE-NUTM2B(FAM22), t(10;17)(q22;p13) :
Transcrit de fusion BCOR-CCNB3 – Enfants plus âgés (8-14ans)
- Diagnostic clinique
- Signes fonctionnels
Asymptomatique
Si compression : douleur, troubles digestifs
- Signes généraux
Fièvre
Perte de poids
Hypertension artérielle
- Signes physiques
-Masse abdominale
Indolore
Contact lombaire
Surface lisse ou bosselée
Consistance ferme ou dure
– Hématurie macroscopique
-Signes de compression : Varicocèle
-Signes de rupture (urgence thérapeutique) : Pâleur, douleur, défense, contracture
-Bandelette urinaire
- Diagnostic paraclinique
- Standards
- Biologie
-NFS, GsRH, hémostase
-Créatininémie, urémie, ionogramme sanguin, ionogramme urinaire
-Calcémie, Catécholamines (sang, urines)
-LDH
-Protidémie
- Imagerie
-Échographie abdomino-pelvienne
Signe de l’éperon
Analyse de la tumeur : Masse solide et ou kystique à échogénicité hétérogène, bien délimitée, avec des zones hypoéchogènes discrètes correspondant à la nécrose, provoquant une distorsion et un déplacement du système collecteur et de la capsule. Apprécier la taille en 3 dimensions.
Rapports vasculaires (Pédicules rénaux, Aorte et ses branches)
Recherche de thrombus (Échographie doppler ++)
Recherche de métastase au niveau du foie ou de l’abdomen
Rein controlatéral
-Radiographie du thorax de face et de profil
Recherche de métastases pulmonaires sous forme de lésions blanches arrondies, souvent situées à la périphérie des poumons.
-TDM TAP avec injection de PDC / TDM Thoracique et IRM Abdomino-pelvienne
Analyse de la tumeur : Masse intrarénale sphérique, hétérogène avec des calcifications et de la graisse. Bien délimitée, avec une atténuation mixte, un léger rehaussement après contraste, des zones kystiques dues à des hémorragies et à la nécrose. Signal faible sur les images pondérées en Hyposignal T1 et hypersignal T2. Apprécier taille dans les 3 dimensions
Anatomie vasculaire
Envahissements
Rein controlatéral
Métastases
Thrombus
– Niveau I: thrombus < 2cm dans VCI
– Niveau II: VCI infra-hépatique
– Niveau III: VCI rétro-hépatique
– Niveau IV: VCI supra-hépatique
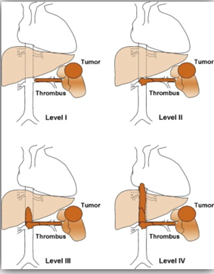
NB : si niveau supérieur du thrombus
- VCI / veines sus-hépatiques : Sd de Budd Chiari aigü
- VCI supra-hépatique : Insuffisance cardiaque aiguë, Embolie pulmonaire
- Diagnostic présomptif de néphroblastome devant :
-Âge (7 mois – 10 ans)
-Rapidité évolutive
-Imagerie typique
- Pas de biopsie, prise en charge thérapeutique sans preuve histologique
- Pas de diagnostic présomptif et indication de biopsie devant :
-Âge > 10 ans
-Infection urinaire
-Biologie atypique : hypercalcémie, Syndrome inflammatoire, élevation des cathécolamines
-Imagerie non typique
Calcifications, adénopathies volumineuses, Parenchyme rénal non visible
-Métastase non habituelle (non pulmonaire, non hépatique)
Procédure biopsie
Voie percutanée postérieure, rétropéritonéale (pas de biopsie transpéritonéale)
Ponction à l’aiguille fine (18 G)
Échoguidée
Contre-indication des biopsies chirurgicales ou avec une aiguille <18G => stade III d’emblée
- Recommandations locales
Biologie
Imagerie : Échographie abdomino-pelvienne, radiographie du thorax F/P et TDM TAP
Même indication de biopsie
- Traitement
- Standards
1) International Society of Pediatric Oncology (SIOP)
- Chirurgie d’emblée
– Nourrissons < 3 mois, adolescents
– Tumeurs kystiques sans composante charnue
– Urgence chirurgicale
Rupture tumorale avec hémodynamique instable
Occlusion réfractaire au traitement médicale
- Chimiothérapie pré-opératoire
-7 mois à 7 ans
-Entre 3 et 6 mois : discussion de l’indication en RCP
-Entre 7 ans et 10 ans : Discussion de l’indication en RCP
Actinomycine-D
Vincristine
Adriamycine
- Chirurgie
-Après 4 semaines de chimiothérapie
-Après 6 semaines de chimiothérapie : métastases, thrombus cave, bilatérales
- Néphrectomie totale élargie (R0)
Abord transversale en débordant la ligne médiane Transpéritonéale
Décollement colo-pariétal et cadre duodénal pour accéder à la VCI
Rester à l’extérieur de la loge rénale
Décoller rein latéralement et en bas
Ligature de l’uretère le plus bas possible
Abord de l’axe aorto-cave de bas en haut
Tumeur G : attention à Artère rénale droite et AMS / Tumeur D : attention à Veine rénale G
Si thrombus VCI infradiafragmatique et infrahépatique non adhérent : extraction par cavotomie ou par cathéter à ballonnet de Fogarty ou de Foley
Isoler pédicule
Ligature artère rénale puis veine rénale
Picking ganglionnaire (> 7 ganglions) : Hilaire, espace aorto-cave, à distance si anormalement large
- Chirurgie conservatrice
-Indications
Sur décision RCP
TW bilatérale ou sur rein unique
TW sur syndrome prédisposant
Conditions
Tumeur unifocale & polaire
Périphérique ou centrale mais à distance des éléments vasculaires
Faible volume (<300mL), stable ou régressive sous CT
Limites
Absence de rupture préopératoire
Nettes avec le parenchyme sain
Sans envahissement pyélo-caliciel
Pas thrombus veineux
Pas d’envahissement LN
Pas d’infiltration organes de voisinage
Préservant > 50% parenchyme rénal sain
Technique
Abord de la loge rénale
Repérage (imagerie 3D, échographie per-opératoire)
Préservation parenchyme rénal
Peu de clampages
Vasodilatateurs
- Compte rendu-post-opératoire
Stade chirurgical
Stade I : La tumeur est réséquée avec une marge claire.
Stade II : La tumeur est macroscopiquement complètement réséquée.Il y a des ganglions abdominaux impliqués (suspectés).
Stade III : La tumeur est incomplètement réséquée ou rompue, ou il y a une extension vasculaire.
Poids de la tumeur
Inspection rein controlatéral si indiqué
Apparence veine rénale, veine cave, capsule tumorale ganglions lymphatiques : (normaux, suspects, paroi infiltrée, thrombose)
Excision complète ou incomplète, ou la biopsie, seront également enregistrées
Autre structure suspecte ou envahie Complications rupture tumorale (majeure/mineure), hémorragie importante (> 50 ml/kg), hypotension, arrêt cardiaque, lésion vasculaire, obstruction de la veine cave inférieure, infarctus intestinal, lésion intestinale, lésion splénique, lésion hépatique
Résection ou biopsie autre organe viscéral (en raison d’une lésion)
- Chimiothérapie et Radiothérapie postopératoire
Fonction du stade local
Fonction des résultats anatomopathologiques
Chimiothérapie
Tous les patients sauf ceux ayant une tumeur de stade I à faible risque.
Actinomycine-D, Vincristine, Adriamycine, Etoposide, cyclophosphamide, Carboplatine
Radiothérapie
Délai : 10 jours suivant la chirurgie
Radiothérapie abdominale totale : histologie de risque intermédiaire ou élevé avec une rupture tumoral majeur préopératoire ou peropératoire, ou dépôts péritonéaux macroscopiques
Radiothérapie locale : Stade III et stade IV
Radiothérapie pulmonaire : métastases pulmonaires persistante après dixième semaine postopératoire.
2)Protocole Children’s Oncology Group (COG, NTWS)
- Chirurgie d’emblée
Devant toutes les tumeurs opérable
Si tumeur très volumineuse : biopsie
- Chimiothérapie pré opératoire
– tumeur non opérable
– thrombose de la veine cave
Doxorubicin, Dactinomycin and Vincristine
- Chimiothérapie adjuvante
Tous les patients
Exception : enfants de moins de 2 ans avec tumeur de stade I à histologie favorable pesant moins de 550 g, ayant des ganglions lymphatiques prélevés et confirmés négatifs.
- Radiothérapie postopératoire
Délai : 10 jours suivant la radiothérapie
Tous les patients stade III, au niveau du lit tumoral
- Recommandations locales
Recommendations de l’International Society of Pediatric Oncology (SIOP)
- Surveillance
Trois premières années : Examen physique et échographie abdominale
Tous les 3 mois pendant la première année
Tous les 6 mois pendant deuxième et troisième année
- Prévention
- Secondaire
-Surveillance des patients à risque génétique : Examen physique et échographie abdominale tous les 3 mois pendant 7 ans.
-Dépistage prénatal : Identification précoce de malformations pouvant passer inaperçu à la naissance et pouvant rentrer dans un cadre syndromique prédisposant.
-Conseil génétiques : Pour les familles avec une histoire de syndromes héréditaires pour identifier les enfants à haut risque.
- Tertiaire
-Suivi post-traitement rigoureux : Suivi régulier après traitement selon les recommandations,
-Réhabilitation et soins palliatifs : Pour les patients ayant survécu au cancer, fournir des services de réhabilitation pour traiter les effets secondaires à long terme du traitement