HEMATOLOGIE
LEUCEMIE LYMPHOIDE CHRONIQUE
LEUCEMIE LYMPHOIDE CHRONIQUE
1-BACKGROUND
DEFINITION
La leucémie lymphoïde chronique est une prolifération monoclonale de lymphocytes B matures au niveau de la moelle osseuse, du sang puis des organes lymphoïdes secondaires (ganglions ; rate).
2-EPIDEMIOLOGIE
- Incidence annuelle :4 sur 100000 habitants
- Environ 4500 nouveaux cas par an en France
- Elle survient dans 90% après 50 ans avec un âge médian de72ans
- La sex-ratio est à prédominance masculine (60% hommes)
3-RAPPELS
C’est une maladie secondaire à la stimulation antigénique :
- Rôle majeur du BCR et sa voie de signalisation (BTK et PI3Kδ), qui contrôlent très finement la prolifération et de survie des lymphocytes B clonales.
- Rôle majeur du BCR et sa voie de signalisation (BTK et PI3Kδ) : 2 sous-types de LLC selon le type d’antigène :
- -LLC-M : Cellules leucémiques dérivés des lymphocytes B ayant rencontré l’antigène de façon T dépendante dans le centre germinatif (plus de la moitié des cas) = Présence des mutations somatiques des IGHV (IGVH : région variable des chaines lourdes d’immunoglobulines)
-LLC-NM : Cellules de LLC qui pourraient être issues de lymphocytes B naïfs ayant rencontré l’antigène de manière T-indépendante = Absence des mutations somatiques des IGHV
- Potentiel prolifératif surajouté, par des pertes ou des mutations des régulateurs classiques de l’apoptose tel que TP53 (Tumor Protein p53) qui sont mutés ou perdus notamment lors des délétions 17p13 et 11q22-23.
- Défaut d’apoptose par la surexpression de la protéine antiapoptique Bcl-2 (B-cell CLL/lymphoma2), Bcl-xl (B-cell lymphoma-extra large) et Mcl-1 (Myeloid cell leukemia 1). Il existe une sous- expression des protéines proapoptiques telles que Bax (BCL2-associated X protein) ou Bcl-xs (B-cell lymphoma-extrasmall).
- Rôle du microenvironnement : (cellules mésenchymateuses, stromales, des niches vasculaires, NLC, cellules folliculaires dendritiques et cellules T). « Si les cellules de LLC présentent une survie prolongée in vivo, elles entrent très rapidement en apoptose in vitro. La co-culture avec des cellules stromales prolonge leur survie ».
- Modulation de la prolifération : Sécrétion de BAFF (B cell activating factor) et APRIL (inducteurs de l’activation et de la prolifération) sécrètent diverses cytokines angiogéniques
- Modulation de la survie (sécrétion de chimiokines dont notamment le sdf1).
4-DIAGNOSTIC C LINIQUE
Circonstances de découverte :
- De façon fortuite, lors d’un hémogramme montrant une hyperlymphocytose sanguine +++
- Signes cliniques : adénopathies superficielles fermes, indolores, non compressives, symétriques et bilatérales, splénomégalie.
- Suite à une complication infectieuse ou auto-immune.
5-DIAGNOSTIC PARACLINIQUE
Biologie :
- Hémogramme : Hyperlymphocytose > 5 G/L, selon les cas une anémie ou thrombopénie
- Frottis Sanguin : petits lymphocytes matures présence d’ombres de Gumprecht sous forme de noyaux éclatés.
- Test Direct à l’anti globuline
Items | 1 point | 0 point |
Ig de surface | Expression faible | Expression forte |
Expression du CD5 | + | – |
Expression du CD23 | + | – |
Expression du CD79b | Faible ou nulle | Forte |
Expression de FMC7 | – | + |
- Immunophénotypage : examen, il permet de déterminer le score de Matutes

6-TRAITEMENT
- Standards
Le traitement standard est fonction de l’algorithme ci-dessous :
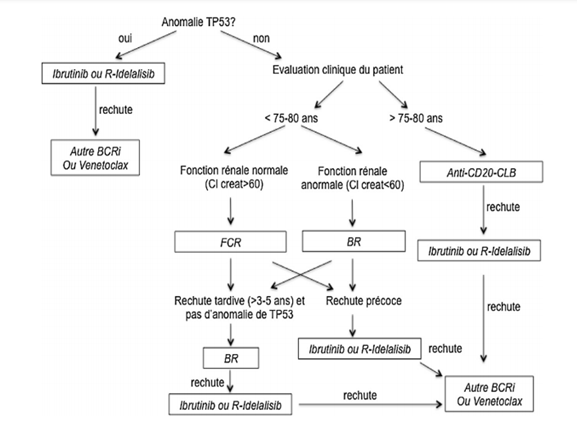
- Recommandations locales
- Chimio-immunothérapie à base de RFC, R-mini-CHOP, R-COP
LYMPHOME DE HODGKIN
LYMPHOME DE HODGKIN
1-BACKGROUNG
A-DEFINITION
Le lymphome de Hodgkin est une prolifération maligne lymphoïde ganglionnaire caractérisée par la présence de cellules de Red Sternberg au sein d’un infiltrante lymphocytaire polymorphe et variable
B-EPIDEMIOLOGIE
- Sujet jeune (25ans) ++++++ / sujet âgé supérieur a 70 ans
- Incidence 2,4sur 100000 habitants par an
- Cancer curable sans séquelle
2-RAPPELS
- ANATOMOPATHOLOGIE
La cellule de Red Sternberg est originaire du centre germinatif des ganglions lymphatiques. Il s’agit d’une grande cellule avec un noyau multilobé, de volumineux et multiples nucléoles et un cytoplasme abondant et clair. Elle se trouve au milieu d’un important infiltrat réactionnel (granulome, richesse ou déplétion en lymphocyte) dont les caractéristiques définissent 4sous types histologiques.
Dans la maladie de hodgkin la cellule tumorale est de nature B meme si les marqueurs de surface de la lignée B ont été perdus (prouve par le réarrangement du gène BCR par PCR)
- CLASSIFICATION HISTOLOGIQUE
La classification OMS distingue 2 types de lymphome de hodgkin :
- Lymphome de hodgkin classique 95% avec 4 sous types histologiques
-Scléro-nodulaire 70%
-Cellularité mixte 20-25%
-Prédominance lymphocytaire 5%
-Déplétion lymphocytaire moins de 5%
- Lymphome hodgkinien à prédominance lymphocytaire nodulaire « LHPL ou paragranulome nodulaire de Poppema et Lennert avec un aspect en popcorn des cellules tumorales. L’immunomarquage est différent de celui de la MH classique.
|
Marqueurs |
HC |
LHPL |
|
CD30 |
+ |
— |
|
CD15 |
+ |
— |
|
CD45 |
— |
+ |
|
EMA |
— |
+/- |
|
Marqueur B CD20 |
— |
+ |
- CLASSIFICATION D’ANN ARBOR
- Stade I : atteinte d’une seule aire ganglionnaire.
- Stade II : atteinte de deux ou plusieurs aires ganglionnaires du même côté du diaphragme.
- Stade III : atteinte ganglionnaire des deux côtés du diaphragme
- Stade IV : atteinte viscérale avec ou sans atteinte ganglionnaire ou atteinte médullaire.
Auxquels il peut être ajouté les lettres :
- E : s’il y a une atteinte viscérale contiguë à une atteinte ganglionnaire
- S : en cas d’atteinte splénique
- A : si pas de signe d’évolutivité B
- B : s’il y a amaigrissement inexpliqué de plus de 10 % du poids du corps en moins de 6 mois ou fièvre inexpliquée >38 °C de plus de 15 jours ou sueurs nocturnes profuses
Les stades I et II constitueront les formes localisées, III et IV celles disséminées.
3-DIAGNOSTIC CLINIQUE
Circonstances de découverte :
- Adénopathies périphériques 80%
- Adénopathies médiatisnales 10%
- Signes généraux 10-20%
EXAMEN CLINIQUE
Circonstances de découverte
Un lymphome peut avoir n’importe quelle localisation et par conséquent, se manifester par des signes cliniques très variés. Cependant, certains tableaux sont plus importants ou fréquents :
- Une ou des adénopathies périphériques,
- Des signes généraux notamment une fièvre au long cours, un amaigrissement, un prurit inexpliqué,
- Des signes compressifs, qui constituent un tableau d’urgence : un syndrome cave supérieur (œdème en pèlerine, turgescence des veines jugulaires, circulation veineuse collatérale thoracique, un syndrome sub-occlusif, un syndrome neurologique de compression médullaire).
- Des atteintes viscérales.
– Signes cliniques
. Signes fonctionnels
Divers symptômes, aspécifiques, peuvent être retrouvés. Ils dépendent très largement de la localisation du ganglion ou de l’organe atteint. Il s’agit en général d’une symptomatologie extra ganglionnaire notamment :
- Digestive (épigastralgies, douleurs abdominales diffuses, troubles du transit)
- Des signes d’atteinte de la sphère ORL (douleur oropharyngée, dysphagie, otalgie, obstruction nasale)
- Des signes neurologiques (paraplégie, syndrome de compression médullaire)
- Des signes cutanés, signes gynécologiques, ophtalmologiques, rénaux, entre autres.
. Signes généraux
Les LNH peuvent se manifester par une altération de l’état général avec amaigrissement, asthénie, anorexie, une fièvre au long cours, des sueurs nocturnes profuses.
Plusieurs échelles sont utilisées pour l’évaluation de l’état général, notamment l’échelle d’activité de l’ECOG, adoptée par l’OMS.
. Signes physiques
Plusieurs tableaux peuvent être rencontrés :
- Des adénopathies superficielles (2/3 des cas) : elles sont fermes, indolores, souvent cervicales et unilatérales, ou diffuses au niveau de plusieurs aires ganglionnaires, de taille supérieure à 2 cm, d’ancienneté supérieure à 1 mois. Elles peuvent s’associer à une splénomégalie.
- Adénopathies médiastinales (20% des cas) associées parfois à un syndrome de la veine cave supérieure.
- Adénopathies rétropéritonéales, mésentériques, pelviennes.
- Une splénomégalie isolée.
- Une hépatomégalie peut aussi être présente, encore moins fréquente.
- Des localisations viscérales qui peuvent être ORL (avec hypertrophie des amygdales, obstruction du cavum), digestives, cutanées (papules infiltrantes multiples, rouges violacées), ophtalmologiques, cérébrales, méningées, osseuses ou épidurales (avec compression médullaire), gonadiques, pulmonaires, thyroïdiennes, entre autres, avec les signes cliniques afférents.
- Des atteintes médullaires peuvent être révélées par une anémie, des infections à répétition, des saignements en rapport avec une thrombopénie.
4-DIAGNOSTIC PARACLINIQUE
– Hémogramme
La réalisation d’un hémogramme peut montrer des anomalies à type d’anémie, de thrombopénie, d’hyperleucocytose avec lymphocytes atypiques au frottis sanguin ou au contraire de leuco neutropénie, d’éosinophilie.
On recherchera attentivement la présence d’un envahissement sanguin éventuel.
L’hémogramme peut toutefois être normal.
– Cytoponction
Il s’agit en général d’un examen d’orientation.
Les prélèvements peuvent se faire sur un ganglion périphérique ou sur un autre site tumoral. La ponction ganglionnaire à l’aiguille fine peut montrer des cellules lymphomateuses ou être peu contributive.
Dans tous les cas, une biopsie ganglionnaire ou d’un autre site suspect s’impose, pour affirmer le diagnostic, en préciser le type histologique et participer au pronostic et à la décision thérapeutique.
– Biopsie ganglionnaire ou tissulaire
L’examen anatomopathologique est indispensable pour poser le diagnostic de LH.
Une biopsie chirurgicale est nécessaire, pour obtenir un fragment ganglionnaire de bonne taille qui permettra :
- Une étude cytologique après apposition ganglionnaire sur lame,
- Une étude histologique standard et des techniques immunologiques,
- La congélation de fragments pour des études immuno-histochimiques plus complètes, voire pour la biologie moléculaire, et éventuellement une étude cytogénétique.
– Immunohistochimie
L’immunohistochimie permet de typer les cellules en détectant des protéines ou d’autres antigènes au moyen d’anticorps, dans des sections de tissu.
- Scanner CTAP : permet évaluer extension
- PET-SCAN : doit être systématique dans le hodgkin
- Biopsie médullaire recherche un envahissement médullaire
5-TRAITEMENT
- Standards
Le traitement dépend du type histologique. Le plus souvent il associe une polychimiothérapie à base de BEACOPP associe ou non à une immunothérapie. Le protocole ABVD peut être aussi utilise en première ligne en fonction du stade et des facteurs pronostics.
- Recommandations locales
- Chimiothérapie a base d’ABVD
6-SURVEILLANCE
La surveillance après traitement des LNH répond au quadruple besoin de :
- Prise en charge des complications immédiates de la chimiothérapie, et éventuellement des autres moyens utilisés,
- Vérification de l’efficacité du traitement à court terme,
- Diagnostic précoce d’une rechute,
- Dépistage d’une potentielle complication tardive du traitement.
L’évaluation de la réponse au traitement repose sur les critères morphologiques de Cheson 1999, établis à partir de la tomodensitométrie, auxquels se substituent les critères de l’International Workshop Criteria (IWC) de 2007 associant réponses morphologique et métabolique (TDM avec injection et TEP). Aux critères de réponse morphologique sont associés des paramètres cliniques, biologiques et anatomopathologiques de la moelle osseuse, pris en compte par l’hématologue pour apprécier la réponse globale.
Les complications tardives du traitement d’un lymphome sont principalement tumorales (tumeurs solides, myélodysplasies et leucémies secondaires), cardiaques (insuffisance cardiaque sévère) et endocrines (infertilité). Le dépistage de ces complications nécessite également une surveillance clinique, biologique et morphologique régulière.
La surveillance clinico-biologique (hémogramme, dosage des LDH et, dans certains cas, de la β2-microglobuline) est primordiale en post-traitement, l’imagerie est effectuée en seconde intention pour le bilan de la rechute, si besoin associée à une TEP. La fréquence des examens de surveillance dépend principalement du type histologique du lymphome.
Leucémies aigües
Leucémies aigües
- Background
- Définition
Les leucémies aigues sont des hhémopathies malignes monoclonales, caractérisées par une infiltration de la moelle osseuse et/ou du sang périphérique par plus de 20% de cellules hématopoïétiques à différenciation bloquée appelées blastes.
Selon leur appartenance à une lignée on distingue : la leucémie aigüe myéloblastique (LAM) de nature myéloïde et la leucémie aiguë lymphoblastique (LAL) de nature lymphoïde.
Possibilité de poser le diagnostic de leucémie aigüe s’il y’a une infiltration d’au moins 10% de blaste avec la présence de certaines anomalies moléculaires caractéristiques (OMS 2022).
- Epidémiologie
La LAL est fréquente chez l’enfant (2 ans – 15 ans) et le sujet après 50 ans.
La LAM est une pathologie de l’adulte, avec un pic après 60 ans.
Il y a une légère prédominance masculine avec un sex- ratio de 1,2
Prévalence hospitalière de 4,8% au service d’hématologie clinique de Dalal Jamm
- Rappels
- Physiologie
Le mécanisme de leucémogénèse s’explique par des mutations successives aboutissant à l’émergence de de gènes oncogènes à l’origine d’un phénotype leucémique. Les cellules leucémiques perdent leur capacité de différenciation et une aptitude à une prolifération accrue et, conférant aux cellules tumorales un avantage de survie lié à un échappement aux règles de mort cellulaire programmée (apoptose).
Sur le plan physiopathologique, en aval, il y a un syndrome tumoral dû à la prolifération maligne qui vont envahir la moelle osseuse entrainant la blastose médullaire, mais également les organes (foie, rate, ganglions etc.) et en amont il y a un syndrome d’insuffisance médullaire
- Diagnostic clinique
- Syndrome tumoral fait :
– Poly adénopathie superficielle, bilatérale et symétrique, intéressant plusieurs aires ganglionnaires, indolore, ferme, sans péri adénite, mobile aux 2 plan, d’apparition récente ;
– Splénomégalie homogène de taille variable à mesurer en cm sous le grill costal ou selon la classification de Hackett ;
– Hépatomégalie ferme, taille variable, indolore, lisse ;
– Douleurs osseuses spontanées diurne et nocturne d’allure inflammatoire ;
– Atteinte dermatologique faite de nodules ou placards fermes enchâssés dans le derme, indolores (leucémide).
– Atteinte neuro méningée qui réalise une méningite blastique, une HTIC et une atteinte des nerfs crâniens (oculo-moteurs, V3 avec anesthésie de la houppe du menton).
– D’atteintes des gonades avec des tumeurs testiculaires ou ovariennes
– D’hypertrophie gingivale (évocatrice de LA monoblastique).
NB : les chloromes sont des tumeurs de nature blastique extra hématopoïétiques
- Les signes d’insuffisance médullaire faits :
- De signes d’anémie : anémie clinique, syndrome anémique,
- De signes hémorragiques : purpura, bulles hémorragique, épistaxis, hématurie, gingivorragie ….
- De signes d’infection pharyngite ulcéronécrotique à fond propre douloureuse (liée à l’agranulocytose), sepsis, ou sepsis sévères lié aux foyers infectieux : ORL, pulmonaires, urogénitales, abcès….
- Diagnostic paraclinique
Numération formule sanguine (NFS) peut montrer :
- Hyperleucocytose ou leucopénie avec neutropénie
- Anémie normo/ macrocytaire et normochrome arégénérative
- Plaquettes : normal ou thrombopénie
- Frottis sanguin montre une blastose
Myélogramme :
Etalement de moelle sur une lame avec coloration au May Grünewald Giemsa montre une prolifération monomorphe de blastes ˃ 20% avec hiatus de maturation. Les autres lignes sont étouffées et rares.
Permet de faire la classification FAB.
Immunophénotypage sur sang périphérique ou médullaire
- Recherche des antigènes cellulaires de surface CD pour classer les leucémies aigues.
- Permet de faire la classification d’Egile : LAL B ou T, LAM ou MIXTE
Cytogénétique :
- Recherche d’anomalie cytogénétique : t (8 ;21), inv 3, inv16, t(15 ;17), t(9 ;22), hyperploidie, caryotype complexe…
- Biologie moléculaire : PML :RAR, NECOM, NMP, CEBPA, BCR : ABL….
- Traitement
- Standards
Chimiothérapie : associe
- Une induction par Cytarabine et daunorubicie
- Suivie d’une consolidation
- Puis d’une allogreffe de cellules souche
- Recommandations locales
- Induction
- Suivie d’une consolidation
- Surveillance
– NFS
– MEDULLOGRAMME
Leucemie Myeloide Chronique (LMC)
Leucemie Myeloide Chronique (LMC)
- Generalités
- Définition
La leucémie myéloïde chronique (LMC) est un syndrome myéloprolifératif clonal dû à la transformation d’une cellule souche pluripotente de la lignée granuleuse et caractérisée sur le plan cytogénétique par la présence de la translocation t(9 ;22).
- Epidémiologie
La leucémie myéloïde chronique (LMC) est la première affection maligne monoclonale de la cellule souche hématopoïétique (CSH) pour laquelle une anomalie chromosomique acquise a été identifiée : le chromosome Philadelphie (Ph). Elle constitue la plus fréquente des syndromes myéloprolifératifs.
Elle représente 15 % de l’ensemble des leucémies de l’adulte et 2 à 5 % des leucémies de l’enfant. Elle affecte les deux sexes, avec une légère prédominance masculine, le sex-ratio Homme / Femme est estimé à 1,3.
Sa fréquence augmente avec l’âge. Elle peut survenir à tous les âges de la vie ; cependant, l’âge médian des patients lors du diagnostic est d’environ 65 ans.
Son incidence est d’environ 1 cas par an pour 100 000 habitants, soit, en France, environ un peu moins de nouveaux cas par an.
- Physiopathologie
La LMC est liée à la survenue dans une cellule souche hématopoïétique d’une anomalie génétique spécifique, acquise, une translocation réciproque et équilibrée touchant les chromosomes 9 et 22 : la translocation (9 ; 22).
Le chromosome 22 est raccourci par l’échange de matériel (der22) et est historiquement appelé «chromosome de Philadelphie» car découvert par des chercheurs de cette ville en 1960.
La conséquence moléculaire de la t(9 ; 22) est la formation d’un gène et d’un transcrit de fusion entre les gènes BCR (situé sur le chromosome 22) et ABL («Abelson», situé sur le chromosome 9).
Le gène ABL code une protéine à activité tyrosine kinase qui est alors délocalisée du noyau vers le cytoplasme et dont l’activité devient permanente par la fusion avec le gène BCR. On parle d’activation constitutive de la kinase, qui interagit avec de nombreuses voies de signalisation. Les conséquences cellulaires de l’activation de la protéine de fusion BCR-ABL sont une prolifération excessive des cellules de la lignée granuleuse, une diminution de l’apoptose et une perte de l’adhérence cellulaire, ce qui explique l’hyperleucocytose et la myélémie
- Diagnostic
2.1 Phase chronique
2.1.1 Circonstance de découverte
La découverte peut être :
- Fortuite : à l’occasion d’une numération- formule sanguine (NFS) de « routine ».
- Signes cliniques : la splénomégalie (SPMG) retrouvée dans plus de 50% des cas de LMC a la phase chronique ou par d’autres signes généraux non spécifiques à la maladie.
- Complications : liées à une thrombopénie tels que des saignements, liées à une thrombocytémie, notamment les complications thrombotiques, ou plus fréquemment, des complications à type de goutte du fait de taux élevés d’acide urique.
2.1.2 Signes Cliniques
Les signes habituellement retrouvés sont
- Une altération de l’état général
- Une satiété rapide ou encore des douleurs abdominales
- Une augmentation du volume de la rate (splénomégalie) est également observée dans la majorité des cas.
2.1.3 signes paracliniques
- Hémogramme
Essentiel au diagnostic, elle révèle :
- Une hyperleucocytose franche supérieure à 20000 voire 50000 /L dans 85% des cas avec 90 à 95% d’éléments granuleux :
- Une polynucléose neutrophile (40-60%)
- Une myélémie constante harmonieuse sans hiatus de différenciation (myéloblastes, promyélocytes, myélocytes, métamyélocytes = 10-50%)
- Une blastose faible inférieure à 5%
- La morphologie de ces éléments granuleux est normale.
Remarque : l’hyperleucocytose peut être franche supérieure à 100 000 GB/L
- Une anémie normochrome normocytaire arégénérative modérée ou absente.
- Une fausse macrocytose
- Une thrombocytose supérieure à 500 G/L
- Myélogramme
Il montre une moelle dont la richesse cellulaire est augmentée, avec une hyperplasie granuleuse marquée et une blastose médullaire inférieure à 10 % en phase chronique. On peut trouver, comme dans le sang, une basophilie, voire une éosinophilie. Les mégacaryocytes sont souvent en nombre augmenté et de petite taille. Inutile pour le diagnostic de LMC, le myélogramme permet cependant de confirmer la phase de la maladie et de réaliser le caryotype initial.
- Le caryotype :
Il est réalisé sur échantillon médullaire, met en évidence dans 95 % des cas la présence du chromosome Philadelphie, classiquement présent dans toutes les cellules. Indispensable au diagnostic, il permet aussi de détecter des anomalies cytogénétiques surajoutées et donc de préciser la phase de la maladie.
- L’hybridation in situ ou FISH :
Elle visualise directement le gène de fusion BCR-ABL sur les noyaux, qu’il y ait translocation visible en cytogénétique ou pas. L’avantage de cette technique est de détecter les remaniements BCR-ABL sans chromosome Philadelphie et d’être plus sensible que le caryotype. Elle ne permet pas, en revanche, de mettre en évidence des anomalies cytogénétiques additionnelles. Cependant, elle peut être utile pour rechercher une délétion du chromosome 9, reconnue comme facteur pronostique péjoratif.
- Biologie moléculaire par PCR
Le critère fondamental du diagnostic est la présence du gène de fusion BCR-ABL détecté par biologie moléculaire. La reverse transcriptase polymérase Chain reaction (RT-PCR) met en évidence le transcrit de fusion Bcr-Abl dans les cellules médullaires ou plus facilement, à partir d’un prélèvement sanguin. Elle permet de définir le sous-type moléculaire produit. Cet examen est aujourd’hui indispensable au diagnostic de LMC. Il peut être réalisé à partir d’un prélèvement sur un simple tube à numération de type éthylène diamine tétra-acétique (EDTA), et même après 36 heures à température ambiante.
- Phase d’accélération
Elle correspond à la transition entre la phase chronique et la phase blastique. Sa durée est de 12 à 18 mois en moyenne.
2.2.1 Signes cliniques
En phase aigüe, l’altération de l’état général est souvent marquée, les douleurs osseuses sont fréquentes et le syndrome tumoral peut comporter, outre la splénomégalie, des adénopathies et une hépatomégalie. Des localisations extra hématopoïétiques sont possibles, en particulier méningées ou des tissus mous (chloromes). Des manifestations en rapport avec l’insuffisance médullaire peuvent être présentes, telles des hémorragies, des infections ou un syndrome anémique marqué.
2.2.2 Signes paracliniques
L’un au moins des critères suivants est présent :
– blastes sanguins ou médullaires entre 15 et 29 %
– blastes + promyélocytes sanguins et médullaires
> 30 % et blastes < 30 %
– basophiles sanguins ≥ 20 %
– plaquettes < 100 G/l en l’absence de toxicité médicamenteuse
2.2.3. Classification OMS 2016 de la phase accélérée
Critères de la phase accélérée de la LMC (OMS 2016) : un ou plusieurs des critères suivants :
- Persistance ou augmentation de la splénomégalie, ne répondant pas au traitement
- Persistante ou augmentation du nombre des leucocytes au-delà de 10 G/L, ne répondant pas au traitement
- Persistance ou augmentation de la thrombocytose (> 1000 G/L), ne répondant pas au traitement
- Persistance d’une thrombopénie < 100 G/L, sans lien avec le traitement
- Présence d’au moins 20% de basophiles dans le sang
- Présence de 10-19% dans le sang et ou la moelle osseuse
- Présence d’anomalies cytogénétiques clonales additionnelles au Ph1, incluant les anomalies de première importance (doublement du Ph1, trisomie 8, isochromosome 17q, trisomie 19), ou un caryotype complexe, ou des anomalies en 3q26.2
- Toute anomalie clonale nouvelle dans des cellules Ph1+ survenant pendant le traitement.
2.3 Phase d’acutisation
2.3.1 Signes cliniques
Elle survient avec un délai médian de 4 ans et se définit par la présence de plus de 20 % de blastes médullaires ou plus de 30 % de blastes et promyélocytes sanguins ou médullaires. Elle s’accompagne en général d’une majoration des signes cliniques d’accélération (altération de l’état général, splénomégalie, anémie, thrombopénie, fibrose médullaire) et parfois d’une symptomatologie propre : fièvre, hépatomégalie, adénopathies et douleurs osseuses. Comme toute leucémie aiguë, elle est possiblement accompagnée d’un syndrome tumoral et de signes d’insuffisance médullaire. Des localisations blastiques extramédullaires peuvent également se voir, notamment une atteinte méningée ou des chloromes des tissus mous.
1.3.2 Signes paracliniques
Classification OMS 2016 de la phase acutisée
- Blastes sanguins ou médullaires ≥20%
- Présence de blastes extramédullaire
- Traitement
- Standards
Le traitement est base sur l’utilisation des inhibiteurs de la Tyrosine Kinase (ITK).
On distingue les ITK de 1er génération (Imatinib), de 2e génération (bosutinib, dasatinib, nilotinib) et de 3e génération (ponatinib).
- Recommandations locales
- Excepte le nilotinib, tous les autres ITK sont disponibles et utilisables en pratiques.
- Surveillance
La LMC est un modèle de suivi hématologique. La surveillance des patients doit être régulière.
Elle associe :
– un examen clinique, avec palpation splénique ;
– une surveillance de l’hémogramme, fréquent au début, pour suivre la diminution puis la disparition de la leucocytose, de la myélémie et des autres anomalies présentes au diagnostic, jusqu’à normalisation complète (réponse hématologique complète) ;
– une surveillance cytogénétique, imposant un caryotype sur moelle tous les six mois, jusqu’à ce que la t(9 ; 22) soit indétectable (réponse cytogénétique complète) ;
– une surveillance moléculaire de la décroissance du taux de transcrit BCRABL, réalisée sur un prélèvement sanguin trimestriel puis semestriel.
Cette surveillance est en principe poursuivie à vie, même lorsque le taux sera indétectable (réponse moléculaire).
Un des enjeux des nouveaux médicaments ciblés oraux est de convaincre le patient de la nécessité d’une observance parfaite, obligatoire pour l’obtention d’une très bonne réponse clinique et biologique.
Lymphome Non Hodgkinien
Lymphome Non Hodgkinien
- Background
- Définition
Les lymphomes sont des hémopathies malignes caractérisées par des proliférations clonales des lymphocytes B, T et ou rarement NK de siège de prédilection ganglionnaire mais pouvant intéresser tous les tissus de l’organisme.
- Epidémiologie
Ils constituent le type d’hémopathie maligne le plus répandu dans le monde. En 2018, 509 590 nouveaux cas de LNH ont été diagnostiqués dans le monde avec une incidence beaucoup plus élevée en Asie et en Europe. En cette même période, 48 587 nouveaux cas ont été diagnostiqués en Afrique soit une incidence de 9,5%. Au Sénégal, les LNH constituent la plus fréquente des hémopathies malignes, avec une augmentation d’incidence remarquable.
- Rappel
– Classification d’Ann Arbor
- Stade I : atteinte d’une seule aire ganglionnaire.
- Stade II : atteinte de deux ou plusieurs aires ganglionnaires du même côté du diaphragme.
- Stade III : atteinte ganglionnaire des deux côtés du diaphragme
- Stade IV : atteinte viscérale avec ou sans atteinte ganglionnaire ou atteinte médullaire.
Auxquels il peut être ajouté les lettres :
- E : s’il y a une atteinte viscérale contiguë à une atteinte ganglionnaire
- S : en cas d’atteinte splénique
- A : si pas de signe d’évolutivité B
- B : s’il y a amaigrissement inexpliqué de plus de 10 % du poids du corps en moins de 6 mois ou fièvre inexpliquée >38 °C de plus de 15 jours ou sueurs nocturnes profuses
Les stades I et II constitueront les formes localisées, III et IV celles disséminées.
- Diagnostic clinique
– Circonstances de découverte
Un lymphome peut avoir n’importe quelle localisation et par conséquent, se manifester par des signes cliniques très variés. Cependant, certains tableaux sont plus importants ou fréquents :
- Une ou des adénopathies périphériques,
- Des signes généraux notamment une fièvre au long cours, un amaigrissement, un prurit inexpliqué,
- Des signes compressifs, qui constituent un tableau d’urgence : un syndrome cave supérieur (œdème en pèlerine, turgescence des veines jugulaires, circulation veineuse collatérale thoracique, un syndrome sub-occlusif, un syndrome neurologique de compression médullaire).
- Des atteintes viscérales.
– Signes cliniques
. Signes fonctionnels
Divers symptômes, aspécifiques, peuvent être retrouvés. Ils dépendent très largement de la localisation du ganglion ou de l’organe atteint. Il s’agit en général d’une symptomatologie extra ganglionnaire notamment :
- Digestive (épigastralgies, douleurs abdominales diffuses, troubles du transit)
- Des signes d’atteinte de la sphère ORL (douleur oropharyngée, dysphagie, otalgie, obstruction nasale)
- Des signes neurologiques (paraplégie, syndrome de compression médullaire)
- Des signes cutanés, signes gynécologiques, ophtalmologiques, rénaux, entre autres.
. Signes généraux
Les LNH peuvent se manifester par une altération de l’état général avec amaigrissement, asthénie, anorexie, une fièvre au long cours, des sueurs nocturnes profuses.
Plusieurs échelles sont utilisées pour l’évaluation de l’état général, notamment l’échelle d’activité de l’ECOG, adoptée par l’OMS.
. Signes physiques
Plusieurs tableaux peuvent être rencontrés :
- Des adénopathies superficielles (2/3 des cas) : elles sont fermes, indolores, souvent cervicales et unilatérales, ou diffuses au niveau de plusieurs aires ganglionnaires, de taille supérieure à 2 cm, d’ancienneté supérieure à 1 mois. Elles peuvent s’associer à une splénomégalie.
- Adénopathies médiastinales (20% des cas) associées parfois à un syndrome de la veine cave supérieure.
- Adénopathies rétropéritonéales, mésentériques, pelviennes.
- Une splénomégalie isolée.
- Une hépatomégalie peut aussi être présente, encore moins fréquente.
- Des localisations viscérales qui peuvent être ORL (avec hypertrophie des amygdales, obstruction du cavum), digestives, cutanées (papules infiltrantes multiples, rouges violacées), ophtalmologiques, cérébrales, méningées, osseuses ou épidurales (avec compression médullaire), gonadiques, pulmonaires, thyroïdiennes, entre autres, avec les signes cliniques afférents.
- Des atteintes médullaires peuvent être révélées par une anémie, des infections à répétition, des saignements en rapport avec une thrombopénie.
Certaines formes histologiques ont des présentations cliniques classiques, qu’il faut connaître :
- Le lymphome de Burkitt : il peut envahir n’importe quel tissu, mais la présentation clinique reste dans la grande majorité des cas stéréotypée. Trois grands types de lymphome de Burkitt sont actuellement identifiés : une forme endémique (liée à EBV), une forme sporadique (moins liée à EBV) et une forme observée chez le patient immunodéprimé.
- Une splénomégalie isolée est évocatrice d’un lymphome splénique de la zone marginale.
- Des symptômes digestifs orientent vers un lymphome du MALT ou un lymphome à cellules du manteau.
- Une atteinte du cavum ou de l’amygdale oriente plutôt vers un lymphome B diffus à grandes cellules avec un risque méningé important.
- Une complication par compression révèle habituellement des lymphomes agressifs (syndrome cave supérieur, compression médullaire, etc.).
- Diagnostic paraclinique
– Hémogramme
La réalisation d’un hémogramme peut montrer des anomalies à type d’anémie, de thrombopénie, d’hyperleucocytose avec lymphocytes atypiques au frottis sanguin ou au contraire de leuco neutropénie, d’éosinophilie.
On recherchera attentivement la présence d’un envahissement sanguin éventuel.
L’hémogramme peut toutefois être normal.
– Cytoponction
Il s’agit en général d’un examen d’orientation.
Les prélèvements peuvent se faire sur un ganglion périphérique ou sur un autre site tumoral. La ponction ganglionnaire à l’aiguille fine peut montrer des cellules lymphomateuses ou être peu contributive.
Dans tous les cas, une biopsie ganglionnaire ou d’un autre site suspect s’impose, pour affirmer le diagnostic, en préciser le type histologique et participer au pronostic et à la décision thérapeutique.
Cependant, dans les lymphomes de Burkitt, l’aspect cytologique est, dans sa forme typique, suffisamment caractéristique pour poser un diagnostic formel : le frottis de cytoponction est massivement envahi par une population monomorphe de cellules lymphoïdes de taille moyenne, régulières, à chromatine de densité intermédiaire, plus ou moins nucléolée et à cytoplasme très basophile et souvent vacuolé. À cette population lymphoïde s’ajoutent de nombreux macrophages à corps tingibles reproduisant l’aspect de « ciel étoilé ».
– Biopsie ganglionnaire ou tissulaire
L’examen anatomopathologique est indispensable pour poser le diagnostic de LNH.
Une biopsie chirurgicale est nécessaire, pour obtenir un fragment ganglionnaire de bonne taille qui permettra :
- Une étude cytologique après apposition ganglionnaire sur lame,
- Une étude histologique standard et des techniques immunologiques,
- La congélation de fragments pour des études immuno-histochimiques plus complètes, voire pour la biologie moléculaire, et éventuellement une étude cytogénétique.
La biopsie ganglionnaire montre une destruction de la structure histologique normale remplacée par des cellules lymphomateuses, précise l’aspect diffus ou nodulaire de la prolifération, ainsi que la taille des cellules qui infiltrent le ganglion.
– Immunohistochimie
L’immunohistochimie permet de typer les cellules en détectant des protéines ou d’autres antigènes au moyen d’anticorps, dans des sections de tissu.
– Immunophénotypage
Il est réalisé soit sur le sang périphérique en phase de dissémination sanguine, soit sur la moelle osseuse, et précise le caractère différencié de la prolifération. Il peut être associé à une étude immunohistochimique et immunocytochimique.
- Traitement
- Standards
Le traitement dépend du type histologique. Le plus souvent il associe une polychimiothérapie à base de CHOP associe à une immunothérapie suivie ou non d’un entretien. Dans certains cas une autogreffe est discutée d’emblée après une première rémission.
- Recommandations locales
- Chimio-immunothérapie
- Autogreffe de moelle osseuse pour certaines formes histologiques
- Surveillance
La surveillance après traitement des LNH répond au quadruple besoin de :
- Prise en charge des complications immédiates de la chimiothérapie, et éventuellement des autres moyens utilisés,
- Vérification de l’efficacité du traitement à court terme,
- Diagnostic précoce d’une rechute,
- Dépistage d’une potentielle complication tardive du traitement.
L’évaluation de la réponse au traitement repose sur les critères morphologiques de Cheson 1999, établis à partir de la tomodensitométrie, auxquels se substituent les critères de l’International Workshop Criteria (IWC) de 2007 associant réponses morphologique et métabolique (TDM avec injection et TEP). Aux critères de réponse morphologique sont associés des paramètres cliniques, biologiques et anatomopathologiques de la moelle osseuse, pris en compte par l’hématologue pour apprécier la réponse globale.
Les complications tardives du traitement d’un lymphome sont principalement tumorales (tumeurs solides, myélodysplasies et leucémies secondaires), cardiaques (insuffisance cardiaque sévère) et endocrines (infertilité). Le dépistage de ces complications nécessite également une surveillance clinique, biologique et morphologique régulière.
La surveillance clinico-biologique (hémogramme, dosage des LDH et, dans certains cas, de la β2-microglobuline) est primordiale en post-traitement, l’imagerie est effectuée en seconde intention pour le bilan de la rechute, si besoin associée à une TEP. La fréquence des examens de surveillance dépend principalement du type histologique du lymphome.
Myélome multiple
Myélome multiple
- Background
- Définition
C’est une hémopathie maligne incurable caractérisée par une prolifération monoclonale de plasmocytes malins dans la moelle osseuse, secrétant ou non une immunoglobuline complète ou incomplète.
- Epidémiologie
Le taux d’incidence du myélome dans le monde est de 4 cas pour 100 000 individus. Selon GLOBOCAN, l’incidence du myélome a augmenté de 10,2% entre 2018 et 2020. Aux Etats-Unis, Le myélome multiple représente 1 % de tous les cancers et environ 10 % de toutes les hémopathies malignes. Chaque année, plus de 32 000 nouveaux cas sont diagnostiqués et près de 13 000 patients décèdent de la maladie. On estime, en France 5 442 le nombre de nouveaux cas recensés en 2018. Au Sénégal, il le deuxième cancer hématologique derrière la LMC avec une prévalence de 25,8% des hémopathies selon une étude réalisée en 2023 à l’hôpital Dalal Jamm. L’âge médian des patients au moment du diagnostic est d’environ 65 ans. Le myélome multiple est légèrement plus fréquent chez les hommes que chez les femmes.
- Diagnostic clinique
La pathologie se manifeste le plus souvent par une symptomatologie osseuse dominée par :
- Douleurs osseuses sont sourdes, avec des paroxysmes, d’aggravation progressive, rebelles, parfois erratiques, permanentes, non soulagées par le repos, diffuses siégeant le plus souvent un niveau des os plats notamment au rachis, au bassin et aux côtes.
- Tuméfactions osseuses de consistance ferme, indolores sont rares et touchent surtout les os plats.
- Fractures pathologiques du bassin, du fémur, de la clavicule : spontanées ou provoquées par un traumatisme minime
L’examen physique est souvent normal mais peut mettre en évidence
- Une douleur à la pression des vertèbres avec un signe de la sonnette positif
- Un syndrome rachidien qui peut aller jusqu’à la compression médullaire lente
- Signes négatifs : absence d’adénopathies, de splénomégalie et d’hépatomégalie
- Diagnostic paraclinique
- Hémogramme : montre le plus souvent une anémie normochrome normocytaire arégénérative
- Frottis sanguin : retrouve une pseudo-agglutination des GR en rouleaux quelques plasmocytes (inconstants)
- Le bilan phosphocalcique montre une hypercalcémie et la phosphorémie normale en l’absence d’une maladie rénale chronique
- Parfois il s’y associe une altération de la fonction rénale
- L’électrophorèse des protéines sériques montre pic d’allure monoclonale à base étroite et à sommet pointu, migrant dans la zone des gammaglobulines, ou des béta globulines. Elle peut montrer aussi une hypogammaglobulinémie.
- L’immunofixation des protéines sériques caractérise l’isotype monoclonal en donnant la chaine lourde et légère de l’immunoglobuline concerné soit Ig G, l’Ig A, Ig D et la chaine légère kappa ou lambda. L’isotype peut être incomplète donnant seulement une chaine légère.
- L’électrophorèse des protéines urinaires met en évidence une protéinurie de type tubulaire dans la majorité des cas ou de type glomérulaire.
- L’immunofixation des protéines urinaires précise le type d’immunoglobuline, qui est
similaire à celle sécrétée dans le sang qui peut être complète ou incomplète - Le myélogramme confirme le diagnostic avec Infiltration plasmocytaire habituellement supérieure à 10 %, faite de plasmocytes souvent dysmorphiques avec des anomalies de taille, un noyau immature nucléolé, de cytoplasmes (inclusions, en ≪flamme ≫ou spumeux, vacuolés déterminant des cellules de Mott).
- La biopsie ostéo-médullaire Indiquée en cas de moelle pauvre et elle confirme l’infiltration plasmocytaire partielle ou diffuse
- La radiologie standard ou la tomodensitométrie osseuse peut montrer des géodes sous forme de lésions lytiques ou lacunes arrondie/ovalaire, de taille variable, disséminées dites à l’emporte-pièce, à limite nette, sans condensation périphérique, localisées au niveau de la voute crânienne, du rachis, du bassin et de l’extrémité proximale des os longs (fémur, humérus) ; une déminéralisation diffuse, surtout au niveau du rachis donnant un aspect en rail : c’est la myélomatose décalcifiante ; des images de fractures des os ; tassements vertébraux cunéiformes ou en galettes des tumeurs osseuses soufflant l’os et érodant le cartilage.
- L’imagerie par résonnance magnétique: donne les mêmes renseignements avec une meilleure sensibilité et des images d’atteinte médullaire notamment l’épidurite.
Le diagnostic repose sur les critères de l’International Myeloma Working Group (IMWG) de 2014 :
- Une plasmocytose médullaire supérieure à 10% ou extra médullaire supérieure à 60%.
- Un plasmocytome qui est une tumeur osseuse plasmocytaire confirmé par l’immunomarquage.
- Et l’un des critères CRAB :
- C : hyperCalcémie
- R : altération de la fonction Rénale
- A : Anémie normochrome normocytaire arégénérative
- B : les lésions osseuses (Bone en anglais)
- Traitement
- Standards
Le traitement repose sur :
- La chimiothérapie avec plusieurs protocoles codifiés par les sociétés savantes qui peut être associées à l’immunothérapie
- L’autogrelle de moelle osseuse consistant à une greffe de moelle ou le donneur et le receveur est la même personne après une chimiothérapie pour diminuer la masse tumorale.
- La radiothérapie palliative à visée antalgique ou décompressive
- Recommandations locales
Dans notre contexte ou plusieurs molécules de premier choix ne sont disponibles ou l’accessibité n’est pas facile pour la population, les protocoles les plus utilisés associent des triplets et d’une autogreffe de moelle osseuse
- Surveillance
- Elle est clinique :
- L’évaluation de la symptomatologie de départ
- La diminution des masses osseuses
- La récupération des déficits neurologiques
- L’identification des effets secondaires des médicaments
- Sur le plan paraclinique
- L’hémogramme, la calcémie, l’albuminémie, la fonction rénale sont réalisés chaque mois
- Ces bilans ajoutés à l’électrophorèse des protéines sériques ou urinaires pour évaluer la gammapathie sont faits selon le schémas thérapeutiques et plus couramment à mi- cure ou à la fin du traitement pour rechercher la réponse immuno- électrophorétique.
- Le myélogramme pour la réponse cytologique et l’immunophénotypage et la recherche de la maladie résiduelle.